Tutorial
In the sample folder are available all input and output files used in this tutorial. Remember to set environment variables before running any application:
export VICTOR_ROOT=/<your_folder>/victor/
export PATH=$PATH:/<your_folder>/victor/bin/
This program calculates phylogenetic trees for the sequences provided as FASTA input. Use
PhylogeneticTrees -h
to show options and default parameters:
Options:
* [--in <name>] Name of input FASTA file
[--verbose] Verbose mode
Distance matrix:
[--global] Needleman-Wunsch-Gotoh global alignment (default)
[--local] Smith-Waterman local alignment
[--freeshift] Free-shift alignment
[--matrix <name>] Custom distance matrix
[-m <name>] Name of substitution matrix file (default = blosum30.dat)
[-o <double>] Open gap penalty (default = 10.00)
[-e <double>] Extension gap penalty (default = 0.10)
Clustering method:
[--upgma] Unweighted Pair Group Method with Arithmetic Mean (default = Neighbor joining)
Output format:
[--out <name>] Name of the Newick tree format output file (default = to screen)
[--rooted] Reroot the tree with 'mid-point' method (NJ only)
The following example show how to build the UPGMA tree of hemoglobin subunit beta from different species:
PhylogeneticTrees --in hem_b.fasta --out upgma.newick --upgma
Default is NJ:
PhylogeneticTrees --in hem_b.fasta --out nj.newick
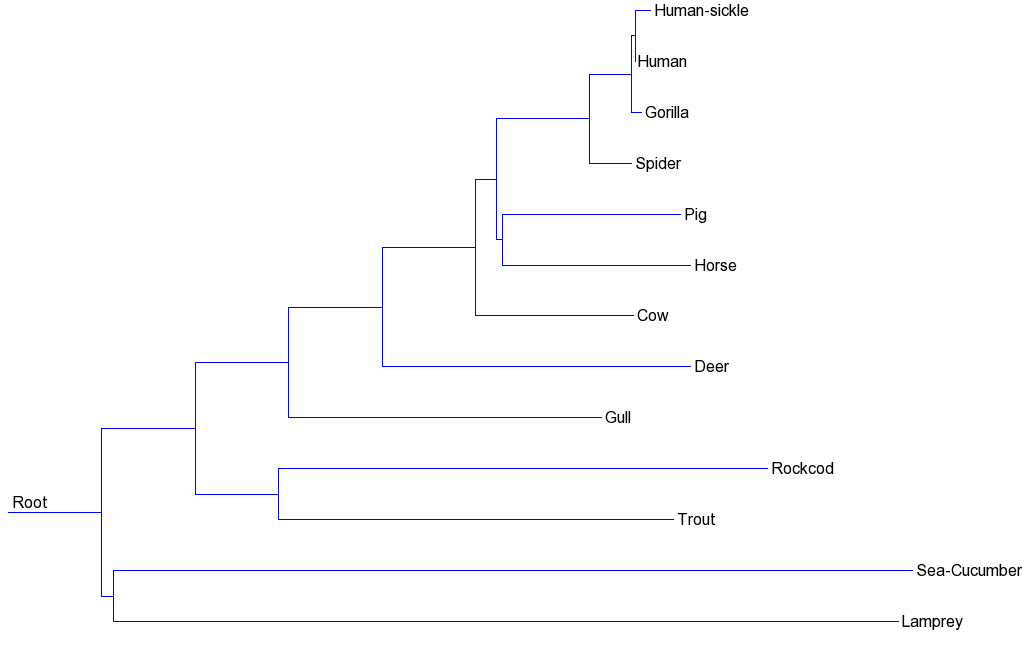
You can use option "--rooted" to get the version of the rooted tree by mid-point method:
PhylogeneticTrees --in hem_b.fasta --out nj.newick --rooted
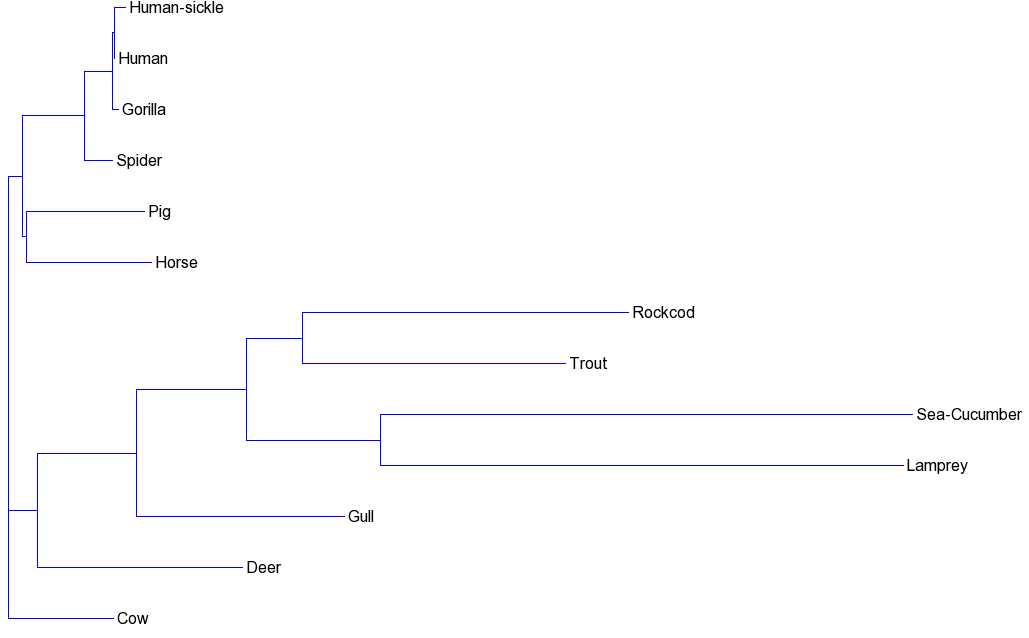
It will produce the files nj.newick (unrooted tree) and rooted_nj.newick (roooted tree). These are the phylogenetic trees obtained and displayed with the Newick Tool from http://trex.uqam.ca/index.php?action=newick&project=trex:
Unrooted NJ tree:
Rooted NJ tree:
This program is an implementation of ClustalW (Thompson, Higgins & Gibson, 1994), one variant of the progressive method for multiple sequence alignment. Use
Msa -h
to show options and default parameters:
Options:
* [--in <name>] Name of input FASTA file
[--verbose] Verbose mode
Distance matrix:
[--global] Needleman-Wunsch-Gotoh global alignment (default)
[--local] Smith-Waterman local alignment
[--freeshift] Free-shift alignment
[--matrix <name>] Custom distance matrix
[-m <name>] Name of substitution matrix file (default = blosum30.dat)
[-o <double>] Open gap penalty (default = 10.00)
[-e <double>] Extension gap penalty (default = 0.10)
Clustering method:
[--upgma] Unweighted Pair Group Method with Arithmetic Mean (default = Neighbor joining)
ClustalW options:
[--cwo <double>] Open gap penalty (default = 10.00)
[--cwe <double>] Extension gap penalty (default = 0.20)
[--cutoff <double>] Cutoff value for divergent sequences (default = 0.4)
[--pam ] PAM substitution matrix series (default = Blosum)
Output format:
[--outNewick <name>] Name of the Newick tree format output file (default = to screen)
[--outMsa <name>] Name of the MSA output FASTA file (default = to screen)
[--clustal] MSA output in clustal format (default = FASTA)
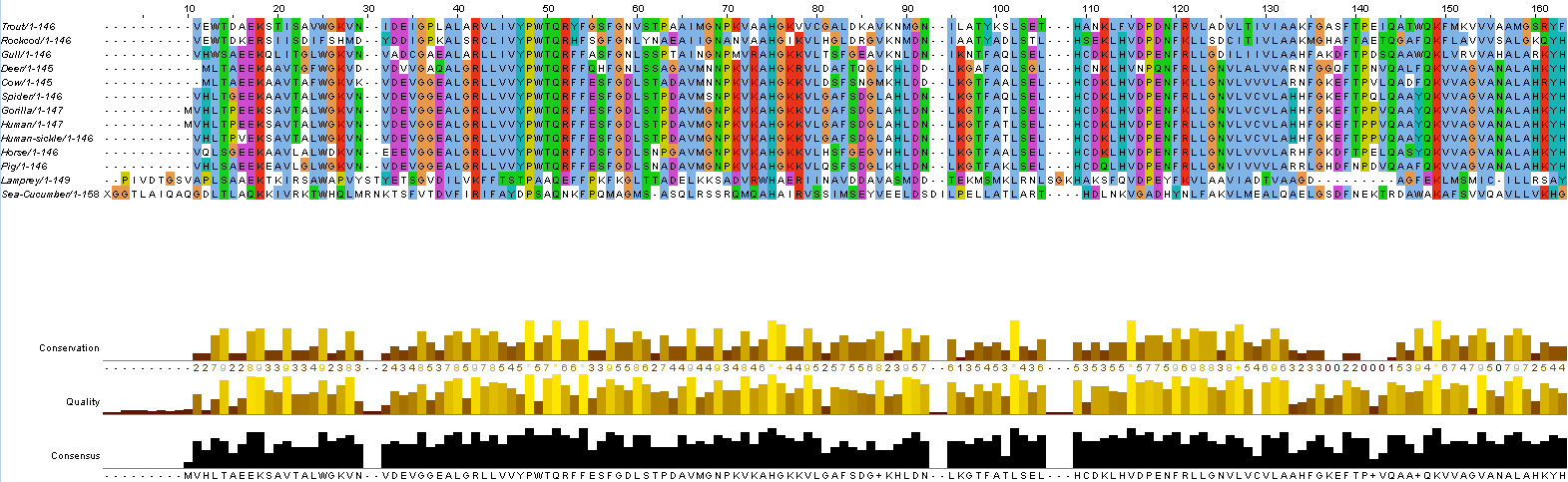
The following example show how to build the MSA of hemoglobin subunit beta from different species:
Msa --in hem_b.fasta --outMsa msa.fasta --outNewick guideTree.newick
This is the result of multiple alignment displayed with Jalview: